
My Journey with the Fruit Fly: Decoding Neurodegeneration in Lysosomal Storage Disease
I am Apurba Das, a recent PhD graduate from the Indian Institute of Science Education and Research Kolkata, India. I have always enjoyed biology classes since school, largely due to my love of nature. I had a childish imagination - as if a tiny version of me could sit inside a cell and watch the processes happening, like watching city traffic, people walking on roads, or workers keeping the city alive. Later, I realised this would require sub-micron visualisation, possible only through microscopes that reveal the cell and its components like the nucleus, lysosome, mitochondria, and proteins. This fascination with the unseen world led me to pursue a PhD - an intuitive choice after my master’s. My core interest was to study human diseases, to understand them better and contribute new knowledge.
My research – Mucopolysaccharidosis Type VII, a Lysosomal Storage Disease
After joining the programme, I was offered a project to study Mucopolysaccharidosis Type VII (MPS VII) using the Drosophila model - yes, those tiny fruit flies you see around in your home. We utilized the power of flies owing to their remarkable advantages: their genes are easy to manipulate and study, they have shorter lifespan which makes ageing research faster, and many of their basic biological processes work in the same way as in humans. Research needs model organisms with genes and processes conserved in humans, as human subjects cannot be used for experiments due to ethical and practical limitations.
MPS VII is an ultra-rare genetic disorder affecting about 1 in 250,000 people, caused by mutations in the β-glucuronidase (β-GUS) gene 1. It belongs to a group of disorders known as lysosomal storage diseases (LSDs), which occur when lysosomes - the organelle responsible for breaking down cellular waste using hydrolytic enzymes fail to degrade materials. When an enzyme or lysosomal protein is missing or dysfunctional, undigested substrates accumulate in the lysosomes, leading to cellular failure and disease.
Our lab generated the first Drosophila model of MPS VII by deleting the CG2135 gene, human homolog of β-GUS. The fly had 70% reduction in β-GUS enzymatic activity and mimicked features like MPS VII patients 2. Mammalian system like mouse models of MPS VII exist, however, their severe disease manifestation creates difficulty for molecular studies. Hence, simpler, and cost-effective models like flies are invaluable.
MPS VII also known as Sly syndrome was first described by Dr. William S. Sly, Professor Emeritus at Saint Louis University, in 1973 (3. He reported that certain patients had distinct facial features, enlarged heads, protruding abdomens, and slow growth. His investigation revealed elevated glycosaminoglycans (GAGs) in urine and presence of granulocytes in blood and bone marrow samples. Dr. Sly identified β-GUS deficiency leads to accumulation of undegraded GAGs which are negatively charged polysaccharides found in connective tissue.
Dr. Sly’s passing on May 26, 2025, marked the loss of a pioneer in lysosomal biology. Beyond MPS VII, he made seminal contributions to understand carbonic anhydrase deficiency, hereditary hemochromatosis, and the discovery of the mannose-6-phosphate receptor - crucial for directing lysosomal proteins to lysosomes alongside Prof. Elizabeth Neufeld. Their discovery later enabled the development of enzyme replacement therapy (ERT) for MPS VII. Prof. Neufeld once reflected on how a small technical oversight had made her miss the receptor in her earlier experiments 4, teaching me that science often hinges on details - a lesson that shaped how I designed my experiments.
To appreciate their contribution, let us take a step back and ask a question, how does a protein “know” it belongs to the mitochondria or lysosomes? The answer is a signal tag (like postal code) that guide proteins to their destinations. Lysosomal enzymes, for instance, are tagged with mannose-6-phosphate in the Golgi apparatus and delivered by vesicles to lysosomes containing the corresponding receptor. These insights not only transformed cell biology but also laid the foundation for targeted drug delivery in modern medicine.

The PhD Problem - Can we correct neurodegenerative symptoms in MPS VII?
When I first learned about MPS VII, I was struck by how devastating it is. The babies with defect in β-GUS gene mostly do not survive birth because of a severe condition that causes their bodies to swell with fluid before delivery also known as non-immune hydrops fetalis. Additionally, those who survive show clinical manifestation like stunted growth, skeletal abnormalities, enlarged liver and spleen, hernias, recurrent infections, cloudy corneas, and joint stiffness. The multi-organ failure is the major cause of mortality, with a median lifespan of MPS VII patients only about 3.5 years.
MPS VII is also a neuropathic disease, affecting cognition and neuromuscular coordination. About 80% of patients show neurodegenerative symptoms - intellectual disability, delayed speech, hearing loss, and restricted movement 1. Neuropathological studies have revealed GAG accumulation, brain vacuolation, and skull enlargement - a system slowly collapsing under its own burden 5,6.
The first and only FDA-approved ERT, Mepsevii™ (vestronidase alfa), developed through Dr. Sly’s collaboration with Ultragenyx biotech company, was approved in 2017 7,8. ERT supplements the missing β-GUS enzyme and benefits MPS VII patients, yet it fails to cross the blood-brain barrier (BBB), leaving neurological symptoms untreated. I noticed that since its first discovery, only a handful of studies have reported on neurodegeneration in MPS VII. However, mechanism of neurodegeneration remains unknown while development of therapy which can restore it, are in preliminary stages. Additionally, ERT can cost around $200,000 annually. This amount is beyond the reach of economically underdeveloped countries like India. This highlights the urgent need for affordable, brain-targeted alternatives.
Drosophila models of MPS VII developed by our group has pitched its importance in the field and was extremely helpful to understand the molecular mechanisms of neuropathology in MPS VII.
Small molecule based therapy - solution for neurodegenerative MPS VII
When I joined the lab, our newly published study on MPS VII fly model showed shorter lifespan, low hatching rates and the flies had drastically reduced ability to climb vertical surfaces known as climbing index. The reduced mobility indicated defect in neuromuscular coordination as our muscle movement depends on the signalling from brain. This prompted us to investigate the tissues, where we found signs of muscle degeneration, loss of dopaminergic neurons, and accumulation of engorged lysosomes, mitochondria, and undegraded proteins in the brain 2. However, the mechanism of development of neurodegeneration in MPS VII was still poorly understood.
We asked a basic question: Why is there accumulation of mitochondria, undegraded proteins, lysosomes in MPS VII brain? Is there any defect in clearance system of the brain? Cells keep themselves clean by removing the unwanted proteins and damaged organelles using pathways like autophagy. Autophagy, a word which has gained popularity after the 2016 Nobel Prize in Physiology or Medicine to Prof. Yoshinori Ohsumi, is explored by number of pharmaceutical companies for its benefit in anti-ageing effect. The recent trend of intermittent fasting is based on the concept that when we fast or limit calories our body breakdown stored fat or glucose to supplement cells. During this process autophagy is activated, where cells breakdown proteins and get rid of worn out organelles like mitochondria and keep our cells especially neurons healthy. The autophagy process starts by formation of a vesicle known as autophagosome which takes up the materials to be broken down, and autophagosome fuses with lysosomes. The lysosome degrades the dumped materials into amino-acids, units of lipids etc which is supplemented to the cell for its use (Fig. 3). Thus, starvation has beneficial effect but beyond a certain threshold our body may not cope with nutritional deficiency.

Accordingly, we first investigated if autophagy is defective in MPS VII flies. We studied them in different age groups and distributed flies into early age 4 days old to late age 45 days old. When the MPS VII flies were deprived of food, we found severe decrease in their lifespan whereas normal flies had longer lifespan indicating they can cope the stress better. These data indicate that MPS VII flies are unable to compensate for nutritional deficiency due to an autophagy defect. I was excited after my first experiment and was happy that my hypothesis got a lead.
Next, I delved deeper and systematically dissected the autophagy process, checking all its steps and found significant reduction in genes and proteins related to autophagy in 30-45 days old MPS VII fly brain 9. An autophagy marker protein known as LC3 or Atg8 was found to be downregulated in MPS VII fly brain. Atg8 is important for formation of the autophagosome, engulfing materials in the autophagosome, and fusing with lysosomes to dump the contents in them. Reduction in its level leads to autophagy defect ultimately triggering cell death in the neural cells. These findings clearly confirmed the underlying reason for neurodegeneration in MPS VII.
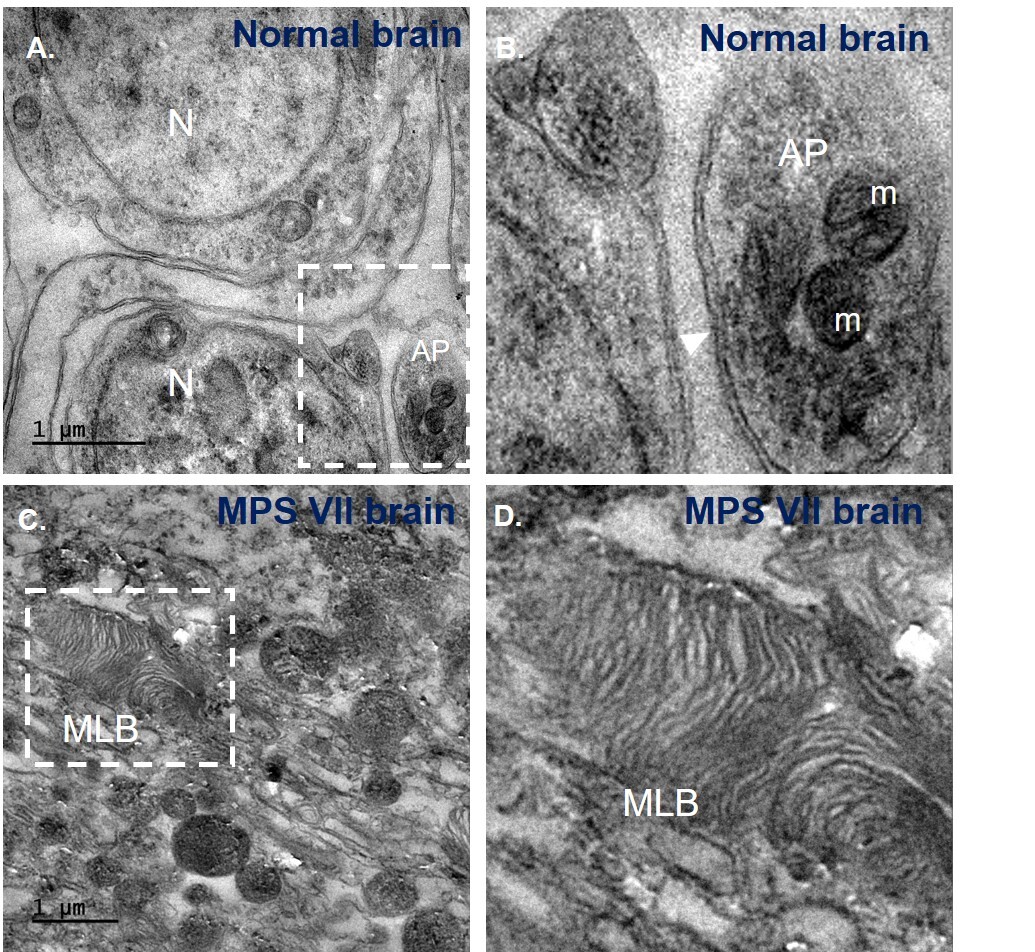
The crucial point in my research was observing the autophagosome in cells under an electron microscope. We found MPS VII had fewer autophagosomes compared to normal brain indicating functional autophagy. Moreover, MPS VII brain had significant presence of multilamellar bodies (MLB) - onion-like structures due to accumulation of undegraded materials in lysosomes (Fig. 4). Reliving my childhood imagination, it was possible to see a nucleus, mitochondria with cristae like I have seen in textbooks, autophagosome - a double membrane structure with mitochondria inside it by using electron microscope. This technique marked an advancement for our lab, as we were finally able to image brain tissues after overcoming several hurdles. This taught me to be persistent and patient, and to trust the process even when things take a long time without much explanation.
The final objective of my project was to find alternative targetable molecules to restore neurodegeneration in MPS VII. For this we turned our focus on using small molecules which can be synthetic or natural derivatives, have low molecular weight and have emerged as therapeutics, especially for neurodegenerative diseases like Alzheimer’s, Parkinson’s, and Huntington’s, etc. They are used due to their versatility, ability to reach intracellular targets, and ease of oral administration, in addition to being cost-effective. We screened a few candidates and found two drugs with the potential to rescue neuropathology by activation of autophagy. We found that trehalose, a non-reducing disaccharide, and resveratrol, a polyphenol extracted from nuts and berries, when orally administered for 30 days, could restore the autophagy defect in the MPS VII brain and improve the climbing ability of the flies.
I also emphasized the idea of finding a drug that can upregulate lysosome biogenesis along with autophagy, so that activated autophagy does not get stalled due to the unavailability of functional lysosomes. This is like a city’s garbage being collected by garbage collectors (autophagy), but if it is not properly recycled or processed (by lysosomes), the city remains dirty. Thus, we looked for factors that can regulate autophagy like a switch. The transcription factor TFEB, also known as the master regulator of lysosome biogenesis, is that switch 10,11. It controls a vast circuit in cells related to autophagy, lysosomes, and the metabolism of lipids and amino acids. When the switch is off, autophagy remains inactive, but under starvation-like stress, it is activated to generate new lysosomes for degrading the cellular “garbage” carried by autophagy.
I found that these drugs can switch on TFEB expression, leading to the initiation of the autophagy and lysosomal gene circuit. My research identified, for the first time in MPS VII, a novel circuit that could serve as an alternative therapeutic approach. Consequently, the clearance of debris was enhanced, and cell death was reduced in the MPS VII fly brain. This restoration of neurodegenerative markers resulted in a notable improvement in the climbing index of MPS VII flies. Thus, my research paved the way for managing neurodegeneration in MPS VII where ERT failed to rescue.
We see how a deficiency of an enzyme (β-GUS) altered the cellular signalling and manifested pathogenesis in MPS VII. Lysosomes being more than a mere suicide bag, can cross-talk with other cellular components and take charge of the processes. We found how lysosome defect caused the imbalance in autophagy and metabolism. Compounds like resveratrol and trehalose, with their ability to cross the blood-brain barrier, offer a promising starting point for such combinatorial strategies. Ultimately, the goal is translational: to connect the molecular understanding of autophagy-lysosomal regulation with therapeutic development. The hope is that by enhancing the cell’s innate capacity for renewal, we may slow or even reverse neurodegeneration not only in MPS VII but across a spectrum of lysosomal and age-related disorders.
Future perspectives and implications
Although MPS VII is rare, the cellular insights it reveals are remarkably universal. Defective clearance and disrupted stress adaptation are shared signatures across major neurodegenerative disorders such as Alzheimer’s, Parkinson’s, and Huntington’s diseases. The activation of autophagy through TFEB illustrates that neuronal survival depends not only on efficient waste disposal but also on the fine-tuning of metabolic balance. These findings highlight that enhancing intrinsic cellular recycling capacity could serve as a valuable complement to enzyme replacement therapies. Moreover, this study underscores the enduring value of model organisms - Drosophila, though simple, continues to illuminate the fundamental biological logic.
Looking ahead, translating these findings into mammalian systems will be essential. I remain deeply curious about how small molecules might restore neuronal health and whether unexplored metabolites or gut-brain interactions could hold keys to crossing the blood-brain barrier. My hope is that this research contributes to the growing efforts to bring real therapeutic hope to patients with rare neurodegenerative disorders.
This PhD journey has been more than an academic pursuit - it has been a lesson in perseverance, curiosity, and gratitude. Behind every experiment were mentors, collaborators, friends, and family who kept the spark alive. Science often begins with wonder, and I aim to carry that same sense of curiosity forward into every new beginning - continuing to ask questions, chase unknowns, and believe in the quiet power of keep learning.

Apurba Das is a recent PhD graduate from Lysosome Biology and Related Disease lab at Department of Biological Sciences, IISER Kolkata, under the guidance of Prof. Rupak Datta. She is interested in understanding neurodegeneration, lysosomal storage disorders, and rare diseases.
References
- Montaño AM, Lock-Hock N, Steiner RD, et al. Clinical course of sly syndrome (mucopolysaccharidosis type VII). J Med Genet. 2016;53(6):403-418
- Bar S, Prasad M, Datta R. Neuromuscular degeneration and locomotor deficit in a Drosophila model of mucopolysaccharidosis VII is attenuated by treatment with resveratrol. DMM Disease Models and Mechanisms. 2018;11(11)
- Sly WS, Quinton BA, McAlister WH, Rimoin DL. Beta glucuronidase deficiency: Report of clinical, radiologic, and biochemical features of a new mucopolysaccharidosis. J Pediatr. 1973;82(2):249-257
- Neufeld EF. From serendipity to therapy. Annu Rev Biochem. 2011;80:1-15
- Irani D, Kim H ‐S, El‐Hibri H, Dutton R V., Beaudet A, Armstrong D. Postmortem observations on β‐glucuronidase deficiency presenting as hydrops fetalis. Ann Neurol. 1983;14(4):486-490
- Vogler C, Levy B, Kyle JW, Sly WS, Williamson J, Whyte MP. Mucopolysaccharidosis VII: postmortem biochemical and pathological findings in a young adult with beta-glucuronidase deficiency. Mod Pathol. 1994;7(1):132-137
- Chen HH, Sawamoto K, Mason RW, et al. Enzyme replacement therapy for mucopolysaccharidoses; past, present, and future. Journal of Human Genetics 2019 64:11. 2019;64(11):1153-1171
- Cadaoas J, Boyle G, Jungles S, et al. Vestronidase alfa: Recombinant human β-glucuronidase as an enzyme replacement therapy for MPS VII. Mol Genet Metab. 2020;130(1):65-76
- Mandal N, Das A, Datta R. Unravelling a mechanistic link between mitophagy defect, mitochondrial malfunction, and apoptotic neurodegeneration in Mucopolysaccharidosis VII. Neurobiol Dis. 2025;206
- Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016;129(13):2475-2481
- Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science (1979). 2011;332(6036):1429-1433